|
Dr Leo Barco. St Richard's Hospital. Chichester. West Sussex. UK
VASCULITIS DE PEQUEñOS VASOS
Vasculitis cutánea de pequeños vasos (VCPV)
Se trata de una vasculitis confinada exclusivamente a la piel. El diagnóstico se realiza por exclusión. Equivaldría a la vasculitis anteriormente conocida como vasculitis leucocitoclástica, alérgica o por hipersensibilidad.
Se caracteriza por una erupción de lesiones con el mismo tiempo de evolución que se resuelven espontáneamente en pocas semanas o meses. Clínicamente se manifiesta como púrpura palpable y no palpable, vesículas y ampollas hemorrágicas o lesiones urticariformes. Se distribuye preferentemente por miembros inferiores y zonas declives o de presión. Los pliegues suelen estar respetados, aunque puede haber afectación de las mucosas. La erupción es generalmente asintomática aunque algunos enfermos refieren picor o quemazón. No existe sintomatología sistémica o es escasa (artralgias leves, cefalea, febrícula, etc.) y no hay evidencia de vasculitis en otros órganos.
Histológicamente encontraremos edema endotelial con necrosis fibrinoide e infiltrado vascular y perivascular neutrofílico con leucocitaclasia (fragmentos nucleares de neutrófilos alrededor y en la pared de los vasos). La inmunofluorescencia directa es positiva en el 50% y se observa depósito granular de IgM, IgG y/o C3 en y alrededor de los vasos dérmicos superficiales.
Las causas potenciales son fundamentalmente los fármacos y las infecciones (por una respuesta inmune anómala que genera el exceso de inmunocomplejos, no se trata de infección directa de los vasos), aunque también se han descrito como desencadenantes algunos alimentos, neoplasias malignas, enfermedades del colágeno y otras enfermedades autoinmunes (enfermedad inflamatoria intestinal, etc.).
Una vez confirmado el diagnóstico, es fundamental descartar, mediante anamnesis exhaustiva por aparatos, exploración física pormenorizada y analítica elemental, la presencia de afectación vasculítica en otros órganos (ver aproximación al paciente con vasculitis).
El tratamiento pasa por eliminar el factor causante si se encuentra y medidas simples como mantener las zonas afectadas a temperatura ambiente (evitar el sol y frío) y reposo relativo junto a elevación de las piernas. Aunque se han empleado los corticoides tópicos, su efectividad no ha sido demostrada. Cuando el proceso se cronifica o en casos extremos con sintomatología importante, se emplea la colchicina. También se ha empleado dapsona (con o sin colchicina). Los corticoides sistémicos pueden emplearse pero se requieren altas dosis ya que de lo contrario son inefectivos. Otros fármacos raramente empleados son: azatioprina, metotrexato, ciclosporina y ciclofosfamida
|

|
|
|

| | | | | | |
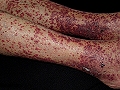
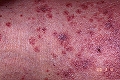
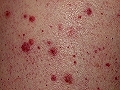
Vasculitis leucocitoclástica
|
Vasculitis leucocitoclástica
|
Vasculitis leucocitoclástica
|
Vasculitis leucocitoclástica
|
Vasculitis leucocitoclástica
|
Púrpura de Schonlein-Henoch (PSH)
Inicialmente descrita como la tétrada de púrpura palpable, artritis, afectación gastro-intestinal y nefritis, en la actualidad se sabe que no siempre se afectan los 4 órganos. En el 75% de los casos aparece entre los 2 y 14 años aunque también se puede presentar en la edad adulta. Es el tipo de vasculitis más común en la infancia y afecta más a los varones.
El cuadro suele comenzar de forma aguda y comúnmente aparece tras una infección de vías respiratorias altas, especialmente por estreptococo. En los adultos también se han implicado los fármacos, en particular los antibióticos del grupo de la penicilina.
Las manifestaciones clínicas más frecuentes son erupción cutánea (95-100%), artralgias (60-80%) y dolor abdominal cólico (85%). El exantema consiste en una erupción macular o maculo-papular, simétrica, de color rojo inicialmente que se torna purpúrica rápidamente. Está constituida por elementos de 1 a 2 mm que por confluencia puede llegar a ser de varios cm. La duración de las lesiones está en torno a las 2 semanas, aunque cerca del 50% de los enfermos presentan brotes recurrentes de púrpura. Se localiza fundamentalmente en piernas y en nalgas aunque en los niños más pequeños también pueden afectarse los brazos y la cara. La afectación renal en forma de micro o macrohematuria es frecuente, pero sólo el 5% evoluciona a enfermedad renal terminal. Se trata de una nefropatía mediada por IgA. La afectación intestinal es variable y suele consistir en dolor cólico agudo con o sin hematoquecia. El sangrado masivo es raro. Cuando se afectan las articulaciones, suelen estar implicadas las rodillas y los codos, pero no suele haber signos inflamatorios. El dolor y la impotencia funcional son lo más característico.
El diagnóstico diferencial en el inicio debe hacerse, en los niños, con el lupus eritematoso sistémico y con la PAN. En el adulto el principal diagnóstico diferencial es con la vasculitis cutánea de vaso pequeño (VCVP) y las vasculitis de vaso pequeño ANCA +.
Los hallazgos de laboratorio son inespecíficos y como más característico en la inmunofluorescencia directa se encuentra depósito de IgA en la pared de los vasos de tamaño pequeño. Este hallazgo no es patognomónico, ya que también se ha detecado en otras enfermedades como el LES, la dermatitis herpetiforme, las endocarditis, el alcoholismo y la enfermedad inflamatoria intestinal.
Para evaluar la extensión de la PSH es imprescindible realizar el estudio de sangre oculta en heces y del sedimento de orina, para descartar la presencia de hematíes y proteínas, ya que el fallo renal es una de las complicaciones más graves. El análisis de orina debe repetirse durante 3-6 meses dado que la afectación renal puede ser diferida. Además del sedimento, debemos solicitar urea y creatinina sanguínea.
Generalmente la enfermedad sigue un curso autolimitado y, si no existe afectación intestinal o renal, el tratamiento es meramente de soporte: asegurar adecuada hidratación, etc. Por supuesto si se encuentra una causa tratable, ésta debe ser eliminada. Existe controversia con respecto al uso de esteroides y sobre su eficacia. Algunos estudios sugieren que el uso temprano puede evitar la evolución de la nefritis o reducir su gravedad pero no son rigurosos. En los casos con nefritis importante parece que puede ser beneficioso el uso concomitante de metilprednisolona, ciclofosfamida y dipiridamol. Los corticoides parecen efectivos para el control del dolor articular y abdominal pero no tienen ningún efecto sobre la púrpura, la duración de la enfermedad o la frecuencia de las recurrencias. Los beneficios de los AINE son escasos y deben evitarse si hay nefritis. Si se dan corticoides hay que asociar ranitidina para reducir el riesgo de sangrado gastro-intestinal.
|

|

|

|
| | | | | | |
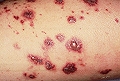
sdme de Schonlein Henöch
|
sdme de Schonlein Henöch
|
sdme de Schonlein Henöch
|
sdme de Schonlein Henöch
| |
Vasculitis crioglobulinémicas:
Las crioglobulinas son inmunoglobulinas que precipitan a bajas temperaturas y su detección en sangre confiere el diagnóstico de crioglobulinemia. El mecanismo patogénico por el cual se produce la vasculitis está ligado a la activación de la cascada del complemento al precipitar las crioglobulinas en la pared del vaso.
Cuando la crioglobulinemia se asocia a malignidad hematológica (mieloma, enfermedad de Waldenstrom) y la inmunoglobulina es monoclonal (ya sea IgG o IgM) y sin actividad factor reumatoide, se denomina Crioglobulinemia de tipo I o simple. Las tipos II y III se conocen como crioglobulinemias mixtas y se acompañan de crioglobulinas con actividad factor reumatoide, que normalmente es de tipo IgM. Si el factor reumatoide es monoclonal se trata de la tipo II y si es policlonal, de la tipo III.
La tipo I generalmente se acompaña de hiperviscosidad más que de vasculitis. Así, la afectación de cara y mucosas, la vasculitis livedoide, la gangrena, el fenómeno de Raynaud y la acrocianosis de los pabellones auriculares inducida por frío son características de la crioglobulinemia simple. Por el contrario, en la tipo II y III lo característico es la presencia de púrpura palpable en extremidades inferiores y en el abdomen, siendo rara la afectación de cara y tronco. También pueden observarse lesiones urticariales.
Entre las causas reconocibles de crioglobulinemia mixta se encuentran las infecciones. La tipo II se asocia especialmente a hepatitis C. Otros virus incriminados son los de la hepatitis A y B, el virus de Epstein-Bar, el citomegalovirus y el VIH. También pueden cursar con crioglobulinemia las enfermedades del colágeno, principalmente el LES, la artritis reumatoide y el síndrome de Sjögren.
Generalmente el enfermo con crioglobulinemia aqueja cansancio y artralgias o artritis, siendo la afectación neurológica y renal un hecho relativamente común. Curiosamente, a nivel cutáneo, sólo entre el 10 y el 30% de los casos refiere agravamiento o aparición de las lesiones con el frío.
Aparte de la analítica solicitada en cualquier vasculitis, es imprescindible solicitar proteinograma y estudio de crioglobulinas (las muestras deben tomarse en frascos que estén a temperatura próxima a los 37 grados). Histológicamente se observa en la tipo I una vasculopatía trombogénica que conduce a la necrosis y en la tipo II y III, una importante vasculitis leucocitoclástica que se extiende por todo el espesor de la dermis. Los precipitados globulosos eosinofílicos en la luz de los vasos inflamados son lo más característico. La IFD muestra depósitos granulares de IgM y C3 en los vasos y en la periferia de los mismos.
En los casos asociados a hepatitis C el tratamiento de elección es el interferón alfa y en los casos asociados a enfermedad hematológica o del colágeno se debe tratar la enfermedad de base. En el resto de casos los corticoides sistémicos se emplean a dosis bajas para el control de la púrpura, las artralgias y la debilidad, y a altas dosis para la enfermedad renal y la neurológica.
|
|
|
|
| | | | | | |
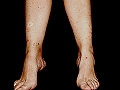
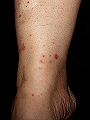
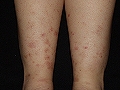
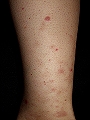
Vasculitis crioglobulinémica
|
Vasculitis crioglobulinémica
|
Vasculitis crioglobulinémica
|
Vasculitis crioglobulinémica
| |
Vasculitis urticarial o urticaria con vasculitis (UV):
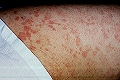
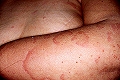
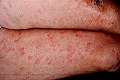
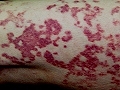
Entre un 5 y un 10% de los pacientes con urticaria crónica presenta UV. Se caracteriza por la presencia de lesiones habonosas que duran en una misma localización más de 24 horas, que duelen o producen quemazón más que prurito y suelen presentar coloración purpúrica que no blanquea a la presión, dejando pigmentación residual en ocasiones.
Existen 2 tipos: normocomplementémica (70-80% de casos) e hipocomplementémica. La primera se puede considerar un subtipo de VCPV y la segunda es relativamente frecuente que se acompañe de enfermedad sistémica (artritis, asma, síntomas gastrointestinales). Algunos de estos últimos enfermos presentan clínica y hallazgos de laboratorio muy parecidos a los del LES.
Para el diagnóstico de UV es fundamental la biopsia cutánea que mostrará infiltrado neutrofílico perivascular e intersticial con signos leves de vasculitis leucocitoclástica.
El tratamiento no está universalmente aceptado. En los casos con poca afectación se pueden intentar los antihistamínicos. La indometacina parece ser beneficiosa en el 50% de los casos. La mayoría responden a los esteroides pero cuando se requiere tratamiento prolongado se prefiere el uso de dapsona (con o sin pentoxifilina), colchicina o hidroxicloroquina.
|
|
|
|
| | | | | | |
Urticaria con vasculitis
|
Urticaria con vasculitis
|
Urticaria con vasculitis
|
Vasculitis urticariforme
| |
|